





Risk of bleeding with non-vitamin K antagonist oral anticoagulants
Bleeding is the main adverse effect when using oral anticoagulants (OACs). The risk of major bleeding with vitamin K antagonists (VKAs) is dependent on the quality of anticoagulation control and estimated to be ∼1.3/100 patients/year in patients with INR 2.0–3.0.1,2 Multiple limitations of VKAs stimulated the development of non-vitamin K antagonist oral anticoagulants (NOACs) acting directly on coagulation factors, including factor FIIa (thrombin) and FXa. Pharmacological properties of the NOACs relevant for bleeding are summarized in the supplement including Supplementary material online, Table S1 and their main effects are summarized in Supplementary material online, Table S2. The risk of bleeding in patients managed with NOACs was properly assessed in the four landmark Phase III trials in patients with atrial fibrillation (AF; Table 1). Major bleeding was the principal safety outcome in RE-LY,3,4 ARISTOTLE,5 and ENGAGE AF.6 In ROCKET-AF, the primary safety endpoint was the composite of major and non-major clinically relevant bleeding.7 The risk of major bleeding was significantly reduced with dabigatran 110 mg b.i.d., apixaban and both doses of edoxaban3–6 while the bleeding rates for dabigatran 150 mg b.i.d. and rivaroxaban were similar to those with warfarin (Table 1).
Risk of bleeding with non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation (rates per 100 patients-years)
| Dabigatran 110 mg b.i.d.3,4 | Dabigatran 150 mg b.i.d.3,4 | Rivaroxaban 20 mg o.d.7 | Apixaban 5 mg b.i.d.5 | Edoxaban 30 mg o.d.6 | Edoxaban 60 mg o.d.6 | |
|---|---|---|---|---|---|---|
| Major bleeding | 2.87 vs. 3.57 | 3.32 vs. 3.57 | 3.60 vs. 3.40 | 2.13 vs. 3.09 | 1.61 vs. 3.43 | 2.75 vs. 3.43 |
| Major plus NMCR | NA | NA | 20.7 vs. 20.3 | 4.07 vs. 6.01 | 7.97 vs. 3.02 | 11.10 vs. 3.02 |
| ICH | 0.23 vs. 0.74 | 0.30 vs. 0.74 | 0.50 vs. 0.70 | 0.33 vs. 0.80 | 0.26 vs. 0.85 | 0.39 vs. 0.85 |
| Major GI bleeding | 1.12 vs. 1.02 | 1.51 vs. 1.02 | 3.20 vs. 2.20 | 0.76 vs. 0.86 | 0.82 vs. 1.23 | 1.51 vs. 1.23 |
| Dabigatran 110 mg b.i.d.3,4 | Dabigatran 150 mg b.i.d.3,4 | Rivaroxaban 20 mg o.d.7 | Apixaban 5 mg b.i.d.5 | Edoxaban 30 mg o.d.6 | Edoxaban 60 mg o.d.6 | |
|---|---|---|---|---|---|---|
| Major bleeding | 2.87 vs. 3.57 | 3.32 vs. 3.57 | 3.60 vs. 3.40 | 2.13 vs. 3.09 | 1.61 vs. 3.43 | 2.75 vs. 3.43 |
| Major plus NMCR | NA | NA | 20.7 vs. 20.3 | 4.07 vs. 6.01 | 7.97 vs. 3.02 | 11.10 vs. 3.02 |
| ICH | 0.23 vs. 0.74 | 0.30 vs. 0.74 | 0.50 vs. 0.70 | 0.33 vs. 0.80 | 0.26 vs. 0.85 | 0.39 vs. 0.85 |
| Major GI bleeding | 1.12 vs. 1.02 | 1.51 vs. 1.02 | 3.20 vs. 2.20 | 0.76 vs. 0.86 | 0.82 vs. 1.23 | 1.51 vs. 1.23 |
These data derive from separate studies comparing each NOAC to VKA; there are no head-to-head studies between NOACs.
b.i.d., twice daily; o.d., once daily; GI, gastrointestinal; ICH, intracranial haemorrhage; NA, not applicable; NMCR, non-major clinically relevant.
Risk of bleeding with non-vitamin K antagonist oral anticoagulants in patients with atrial fibrillation (rates per 100 patients-years)
| Dabigatran 110 mg b.i.d.3,4 | Dabigatran 150 mg b.i.d.3,4 | Rivaroxaban 20 mg o.d.7 | Apixaban 5 mg b.i.d.5 | Edoxaban 30 mg o.d.6 | Edoxaban 60 mg o.d.6 | |
|---|---|---|---|---|---|---|
| Major bleeding | 2.87 vs. 3.57 | 3.32 vs. 3.57 | 3.60 vs. 3.40 | 2.13 vs. 3.09 | 1.61 vs. 3.43 | 2.75 vs. 3.43 |
| Major plus NMCR | NA | NA | 20.7 vs. 20.3 | 4.07 vs. 6.01 | 7.97 vs. 3.02 | 11.10 vs. 3.02 |
| ICH | 0.23 vs. 0.74 | 0.30 vs. 0.74 | 0.50 vs. 0.70 | 0.33 vs. 0.80 | 0.26 vs. 0.85 | 0.39 vs. 0.85 |
| Major GI bleeding | 1.12 vs. 1.02 | 1.51 vs. 1.02 | 3.20 vs. 2.20 | 0.76 vs. 0.86 | 0.82 vs. 1.23 | 1.51 vs. 1.23 |
| Dabigatran 110 mg b.i.d.3,4 | Dabigatran 150 mg b.i.d.3,4 | Rivaroxaban 20 mg o.d.7 | Apixaban 5 mg b.i.d.5 | Edoxaban 30 mg o.d.6 | Edoxaban 60 mg o.d.6 | |
|---|---|---|---|---|---|---|
| Major bleeding | 2.87 vs. 3.57 | 3.32 vs. 3.57 | 3.60 vs. 3.40 | 2.13 vs. 3.09 | 1.61 vs. 3.43 | 2.75 vs. 3.43 |
| Major plus NMCR | NA | NA | 20.7 vs. 20.3 | 4.07 vs. 6.01 | 7.97 vs. 3.02 | 11.10 vs. 3.02 |
| ICH | 0.23 vs. 0.74 | 0.30 vs. 0.74 | 0.50 vs. 0.70 | 0.33 vs. 0.80 | 0.26 vs. 0.85 | 0.39 vs. 0.85 |
| Major GI bleeding | 1.12 vs. 1.02 | 1.51 vs. 1.02 | 3.20 vs. 2.20 | 0.76 vs. 0.86 | 0.82 vs. 1.23 | 1.51 vs. 1.23 |
These data derive from separate studies comparing each NOAC to VKA; there are no head-to-head studies between NOACs.
b.i.d., twice daily; o.d., once daily; GI, gastrointestinal; ICH, intracranial haemorrhage; NA, not applicable; NMCR, non-major clinically relevant.
Gastrointestinal (GI) bleeding accounts for ∼90% of major extracranial haemorrhages in patients with AF receiving VKAs.8 With the NOACs, dabigatran 150 mg b.i.d., rivaroxaban, and edoxaban 60 mg o.d. significantly increased rates of GI bleeding (∼1.5-fold) compared with warfarin.3,6,7 While rivaroxaban increases upper and lower GI bleeding to a similar extent, dabigatran 150 mg b.i.d. prominently increased lower GI bleeding.9 Apixaban was associated with GI bleeding rates comparable with those with warfarin.5 Edoxaban 30 mg o.d. demonstrated significantly lower rates of GI bleeding compared with warfarin (Table 1).6
Intracranial haemorrhage (ICH) is the major concern in patients on OACs and this is the field where NOACs demonstrated major clinical improvement (Table 1). In the four trials, the hazard ratio for ICH was between 0.31 and 0.64, with meta-analysis showing a hazard ratio of 0.48 overall.10 Furthermore, the sequelae of ICH may be less severe when it has occurred during NOAC treatment.11 Major bleeding and ICH rates in ‘real-world’ post-marketing data for NOACs also seems to be generally reassuring, although GI bleeding rates appear to be higher for dabigatran (150 mg b.i.d.) compared with warfarin.12–14
While this paper focuses on the reversal of the effects of NOACs due to an emergency situation, prevention of bleeding remains of utmost importance. The risk of bleeding may be estimated using the well-validated HAS-BLED score.15 A high HAS-BLED score is there to ‘flag up’ patients at higher risk of bleeding for more careful review and follow-up, as well as attention to the potentially reversible risk factors for bleeding, such as uncontrolled hypertension (systolic blood pressure >160 mmHg), labile INRs (for VKA user), concomitant use of aspirin/NSAIDs, alcohol excess. Among AF patients, the HAS-BLED score has been validated to be predictive of bleeding with VKAs, aspirin, and NOACs.16–19
The need to reverse the effect of NOACs occurs in case of life-threatening bleeding, bleeding which cannot be controlled by withdrawal of OAC and usual care, or medical emergencies other than bleeding requiring immediate surgery (Figure 1). Intracranial haemorrhage, with the highest likelihood for fatal outcome, is one of the primary aims for reversal agents. A timely administration of reversal agents is likely to be of major relevance for an improved clinical outcome in case of ICH. However, bleeding at other sites may also be life-threatening and risk stratification is more difficult but essential for these bleeding events. A subanalysis of the RE-LY trial showed that attempts to reverse the effect of dabigatran with clotting factor concentrates (CFCs) were observed in <2% of all major bleeding events.20 Despite the lack of a specific reversal agent, patients in the NOAC arm showed a trend for reduced mortality associated with major bleeding compared with VKA, most likely due to a shorter half-life and a lower rate of ICH.20,21 At the present time, available guidance for reversal of uncontrolled bleeding in patients treated with NOACs is based on expert opinion rather than on clinical trials.22–25
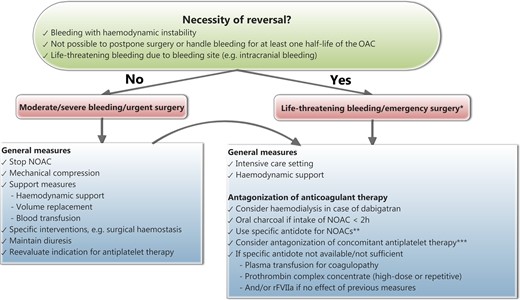
Practical management of the reversal of non-vitamin K antagonist oral anticoagulants. *: requiring normal haemostasis (for other conditions than bleeding); **: when approved by respective regulatory authorities; idarucizumab has been approved for reversal of the effect of dabigatran by the European Medicines Agency and the U.S. Food and Drug Administration; ***: antagonization of concomitant anti-platelet therapy: • Reconsider combination therapy and hold off anti-platelet therapy until safe/appropriate to restart. • Consider platelet transfusion if receiving irreversible anti-platelet drug (aspirin, clopidogrel, and prasugrel) or thrombocytopenia. • Consider specific antidote (MEDI2452) if receiving ticagrelor and when antidote approved by respective regulatory authorities and available.
Existing reversal strategies for non-vitamin K antagonist oral anticoagulants
As there are no specific antidotes available so far, several therapeutic interventions have been used, including activated charcoal, haemodialysis, activated charcoal haemoperfusion, fresh frozen plasma (FFP), and pro-haemostatic agents.22–25 This section will focus on the potential of the pro-haemostatic CFCs for reversal of NOAC effects encompassing pro-thrombin complex concentrates (PCCs) and recombinant (r)FVIIa (Novoseven®, NiaStase©). The ESC Guidelines for the management of AF have recommended considering these CFCs in case of very severe bleeding.26 Pro-thrombin complex concentrates, developed for reversal of VKAs, are lyophilized concentrates of vitamin K-dependent coagulation factors available as: (i) non-activated three-factor PCCs (Profilnine® SD, Bebulin® VH) containing similar amounts of factors II, IX, and X, and low factor VII levels; (ii) non-activated four-factor PCCs (Beriplex® P/N, Kanokad®, Octaplex®, Kaskadil®, Cofact®), containing adequate levels of factors II, VII, IX, and X, with varying amounts of heparin and proteins C and S to reduce thrombogenicity; and (iii) activated four-factor PCC (FEIBA®) containing factors II, IX, X, and protein C, mainly in non-activated forms, and factor VII mainly in the activated form. rFVIIa is a potent general haemostatic agent.27,28 Fresh frozen plasmas containing normal levels of all coagulation factors have practical constraints for the rapid reversal of OACs. A limitation for the use of PCCs is an absolute increase of thromboembolic events of >1%.29,30 The increase of thromboembolic events associated with the use of rFVIIa may be even higher.31,32
As the effect of pro-haemostatic agents on NOACs has not been tested in clinical trials so far, our evidence is based on surrogate endpoints. In comparison, the effect of PCCs on the reversal of VKA effects has been evaluated in patients with major bleeding or requiring urgent surgery. However, this evidence is likewise based on the effect on coagulation tests in small, mostly uncontrolled, retrospective,33 and prospective clinical trials.34–37 In Supplementary material online, surrogate endpoints for measuring the reversal of NOAC effects with CFCs are critically appraised. Supplementary material online, Table S3 gives a detailed description of the available data for reversal of NOAC effects with CFCs based on normalization of coagulation tests, ex vivo thrombus generation (TG) or animal bleeding models. In summary, the evidence supporting the efficacy and safety of CFCs in case of severe or life-threatening bleeding is based on laboratory or animal studies, case reports and a Phase I trial using punch biopsies38 but not on well-designed clinical trials so far. The results are contradictory with regard to an overall reversal of NOAC effects by CFCs and the preference of one specific CFC. Furthermore, they are confusing due to the lack of correlation between the correction of routine laboratory tests, TG, and bleeding in animal models. In particular, animal data suggest that a high dose of CFCs (e.g. 50 IU/kg of four-factor PCC) is necessary for a sufficient reversal effect. Limited animal data suggest that CFCs may ameliorate the effect of NOACs in case of ICH. An important drawback of the use of PCCs and rFVIIa may be an increased risk of thromboembolic complications.29–32
Pending the availability of robust clinical evidence of a beneficial effect, if immediate haemostatic support is required due to a life-threatening situation, the choice of CFCs may be based on its least pro-thrombotic effect, favouring PCCs over rFVIIa. If a four-factor PCC is used, a high dose or repeated dose is recommended. Finally, the choice of a CFC must be individually assessed according to drug availability and experience of the centre, institutional transfusion guidelines and available consensus among cardiologists, haemostasis experts, and emergency physicians. Although pro-haemostatic agents may also limit the extent of bleeding in patients with ICH, their effect on mortality and disability is unknown.39
The potential benefit of specific reversal agents for non-vitamin K antagonist oral anticoagulants
A number of new reversal agents are under development, including monoclonal antibodies, decoy receptors, and small molecules with high affinity for NOACs (Table 2). These agents aim to specifically, completely, and rapidly reverse the effect of NOACs (Figure 2). Another perceived advantage of these reversal agents is the lack of a pro-thrombotic effect of the agents themselves. However, the sole interruption of anticoagulation therapy may be associated with an increased risk of thromboembolic events. Accordingly, in Phase III trials for AF, an increased thromboembolic risk has been observed at the end of the study after study treatment has been stopped.7 Despite the assumed high efficiency of specific reversal agents, their effectiveness in preventing clinical events needs to be determined. Specifically, life-threatening situations with impairment of renal and liver function will constitute a challenge for improvement of survival by specific reversal of NOAC effects.
New reversal agents for non-vitamin K antagonist oral anticoagulants
| Idarucizumab | Andexanet | Ciraparantag (PER977) | |
|---|---|---|---|
| Target | Dabigatran | Oral direct factor Xa-inhibitors, low-molecular-weight heparins and fondaparinux | Oral direct factor Xa and IIa inhibitors, low-molecular-weight heparins, un-fractionated heparin and fondaparinux |
| Structure | Humanized Fab fragment | Human rFXa variant | Synthetic small molecule |
| Immediate onset of reversal (<10 min) | Yes | Yes | Yes |
| Duration of effect | (12 to) 24 h | 2 h | 24 h |
| Re-administration possible | Yes, after 24 h | Unknown | Currently tested (NCT02207257) |
| Tested in healthy volunteers | Yes (NCT020287809) | Yes | Yes (NCT01826266, NCT02207257) |
| Elderly | Yes | Yes (NCT022207725) | No |
| Renally impaired | Yes | No | No |
| Tested in patients | Successful reversal of the specific effect of dabigatran (NCT02104947) | Ongoing (NCT02329327) | No |
| Pro-coagulation signals | No | Decrease of tissue factor pathway inhibitor activity | No |
| Idarucizumab | Andexanet | Ciraparantag (PER977) | |
|---|---|---|---|
| Target | Dabigatran | Oral direct factor Xa-inhibitors, low-molecular-weight heparins and fondaparinux | Oral direct factor Xa and IIa inhibitors, low-molecular-weight heparins, un-fractionated heparin and fondaparinux |
| Structure | Humanized Fab fragment | Human rFXa variant | Synthetic small molecule |
| Immediate onset of reversal (<10 min) | Yes | Yes | Yes |
| Duration of effect | (12 to) 24 h | 2 h | 24 h |
| Re-administration possible | Yes, after 24 h | Unknown | Currently tested (NCT02207257) |
| Tested in healthy volunteers | Yes (NCT020287809) | Yes | Yes (NCT01826266, NCT02207257) |
| Elderly | Yes | Yes (NCT022207725) | No |
| Renally impaired | Yes | No | No |
| Tested in patients | Successful reversal of the specific effect of dabigatran (NCT02104947) | Ongoing (NCT02329327) | No |
| Pro-coagulation signals | No | Decrease of tissue factor pathway inhibitor activity | No |
The clinical development program for Ciraparantag is currently at the Phase II stage, it is not possible to comment on the dose that will be studied in the Phase III trial and which is therefore more likely to be approved for clinical use.
New reversal agents for non-vitamin K antagonist oral anticoagulants
| Idarucizumab | Andexanet | Ciraparantag (PER977) | |
|---|---|---|---|
| Target | Dabigatran | Oral direct factor Xa-inhibitors, low-molecular-weight heparins and fondaparinux | Oral direct factor Xa and IIa inhibitors, low-molecular-weight heparins, un-fractionated heparin and fondaparinux |
| Structure | Humanized Fab fragment | Human rFXa variant | Synthetic small molecule |
| Immediate onset of reversal (<10 min) | Yes | Yes | Yes |
| Duration of effect | (12 to) 24 h | 2 h | 24 h |
| Re-administration possible | Yes, after 24 h | Unknown | Currently tested (NCT02207257) |
| Tested in healthy volunteers | Yes (NCT020287809) | Yes | Yes (NCT01826266, NCT02207257) |
| Elderly | Yes | Yes (NCT022207725) | No |
| Renally impaired | Yes | No | No |
| Tested in patients | Successful reversal of the specific effect of dabigatran (NCT02104947) | Ongoing (NCT02329327) | No |
| Pro-coagulation signals | No | Decrease of tissue factor pathway inhibitor activity | No |
| Idarucizumab | Andexanet | Ciraparantag (PER977) | |
|---|---|---|---|
| Target | Dabigatran | Oral direct factor Xa-inhibitors, low-molecular-weight heparins and fondaparinux | Oral direct factor Xa and IIa inhibitors, low-molecular-weight heparins, un-fractionated heparin and fondaparinux |
| Structure | Humanized Fab fragment | Human rFXa variant | Synthetic small molecule |
| Immediate onset of reversal (<10 min) | Yes | Yes | Yes |
| Duration of effect | (12 to) 24 h | 2 h | 24 h |
| Re-administration possible | Yes, after 24 h | Unknown | Currently tested (NCT02207257) |
| Tested in healthy volunteers | Yes (NCT020287809) | Yes | Yes (NCT01826266, NCT02207257) |
| Elderly | Yes | Yes (NCT022207725) | No |
| Renally impaired | Yes | No | No |
| Tested in patients | Successful reversal of the specific effect of dabigatran (NCT02104947) | Ongoing (NCT02329327) | No |
| Pro-coagulation signals | No | Decrease of tissue factor pathway inhibitor activity | No |
The clinical development program for Ciraparantag is currently at the Phase II stage, it is not possible to comment on the dose that will be studied in the Phase III trial and which is therefore more likely to be approved for clinical use.
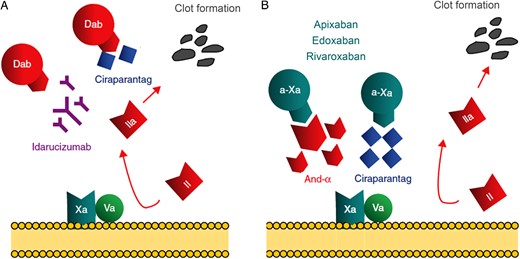
Mechanism of non-vitamin K antagonist oral anticoagulants and their antidotes adapted from Enriquez et al.63 The prothrombinase complex, consisting of FXa and FIIa, catalyses the conversion of pro-thrombin (II) to thrombin (IIa), leading to fibrin generation and clot formation. The effect of the thrombin inhibitor dabigatran (dab) may be reversed by idarucizumab, a humanized antibody fragment against dabigatran OR ciraparantag, a small synthetic molecule competitively binding the non-vitamin K antagonist oral anticoagulants (A). The effect of the FXa inhibitors (a-Xa) apixaban, edoxaban and rivaroxaban may be reversed by andexanet alpha, a modified inactive recombinant FXa competitively binding circulating FXa inhibitors OR ciraparantag (B).
Idarucizumab, an antibody fragment targeting dabigatran
Idarucizumab (BI 655075) is a humanized, highly selective, and specific monoclonal antibody fragment (fab) which binds dabigatran with high affinity (dissociation constant, KD, 2.1 pM), i.e. ∼350 times stronger than for thrombin. It rapidly (<5 min) and completely inhibited the anticoagulant activity of dabigatran in ex vivo human clotting tests (half maximal inhibitory concentration [IC50] of 2–5 nM) as well as in in vivo models, exerting a sustained effect for up to 6 h after intravenous (i.v.) injection.40 However, idarucizumab has no direct activity on coagulation or platelet aggregation.40,41 In Phase I placebo-controlled studies, ecarin clotting time (ECT) and diluted thrombin time (dTT) were significantly prolonged with dabigatran and reversed to control levels after i.v. dosing with idarucizumab (1, 2, 4, and 5 + 2.5 g) in 47 men.42 It was found to be safe and effective, in that dTT was almost immediately corrected.41,42 For the 1 g dose, there was partial return of dabigatran-induced anticoagulation accompanied by an increase of unbound dabigatran concentrations ∼2–4 h after i.v. infusion.42,43 Additionally, idarucizumab reversed dabigatran-induced inhibition of wound site fibrin formation.44 In another 46 male and female volunteers, 2.5–5 g of idarucizumab resulted in immediate, complete, and sustained reversal of dabigatran-induced anticoagulation. The reversal effect was consistent for elderly and renally impaired subjects [creatinine clearance (CrCl) 30 to <90 mL/min).43 Dabigatran anticoagulation could be re-established 24 h after idarucizumab dosing.
An uncontrolled Phase III study (RE-VERSE AD) will assess for the reversal of the anticoagulant effects of dabigatran by the i.v. administration of 5 g of idarucizumab in dabigatran-treated patients, who have uncontrolled bleeding or require emergency surgery or procedures (NCT0210494).45 Up to 300 dabigatran-treated patients with uncontrollable overt bleeding in need of reversal as judged by the attending physician (group A), OR in need of emergency surgery or procedures within the first 4 h (group B) will be enrolled. Patients with bleeding manageable conservatively (group A) or requiring surgery where the risk of uncontrolled or unmanageable bleeding is low (group B) are excluded. The primary endpoint is the maximum reversal of anticoagulant effect of dabigatran in the first 4 h measured with a specific coagulation test (dTT or ECT). A first interim analysis has been performed in 90 patients (A: n = 51, B: n = 39, median age 76.5 years, median CrCl 58 mL/min). The most common bleeding events in group A were GI bleeding (n = 20) and ICH (n = 18). Overall, 22 patients were excluded from further analysis as they already had normal coagulation tests at baseline. Idarucizumab rapidly and completely reversed the anticoagulant activity of dabigatran in 88–98% of patients.46 In parallel, the concentration of unbound dabigatran was reduced to a level at or near the lower limit of quantification. In few patients, a rebound of the effect of idarucizumab was observed after 12 h (n = 6) and 24 h (n = 16), probably due to redistribution of extravascular dabigatran into the intravascular compartment. These results confirm successful reversal of the specific effect of dabigatran in critically ill patients who suffer uncontrollable bleeding or who require emergency surgery while on dabigatran. With regard to clinical bleeding, haemostasis was restored after a median of 11.4 h in group A. In group B, normal intraoperative haemostasis was observed in 92% of patients. However, this normalization of haemostasis did not prevent 18 deaths. One thrombotic event was observed within 72 h after administration of idarucizumab and another four events after 72 h. A controlled design against placebo or CFCs would perhaps have been better, to assess the impact of the reversal on the clinical course of patients. But, given the clinical type of the RE-VERSE AD trial patients, such a design presents major ethical challenges. The interim analysis of the RE-VERSE AD trial led to the approval of idarucizumab by the European Medicines Agency and the U.S. Food and Drug Administration.
Andexanet (PRT064445), a truncated human recombinant FXa
Andexanet alpha (AnXa; PRT064445), a truncated human recombinant FXa, is catalytically inactive and lacks the membrane-binding domain of native FXa but retains the ability of native FXa to bind direct FXa inhibitors as well as low-molecular-weight heparin-activated antithrombin III with high-affinity (mean KD 0.5–1.5 nM for direct FXa inhibitors).47 It acts as a decoy receptor and competes with native FXa, thereby reversing the anticoagulant effects of all FXa inhibitors. AnXa dose-dependently reversed the inhibition of FXa activity by FXa inhibitors and corrected clot formation ex vivo.47 In animal bleeding models, it reduced blood loss and restored haemostasis after pretreatment with rivaroxaban as well as indirect FXa inhibitors (low-molecular-weight heparins and fondaparinux). The effect of AnXa correlated with the reduction in free FXa inhibitor plasma levels and peak anti-FXa activity. However, AnXa did not interfere with normal FXa function in haemostasis and did not have anticoagulant activity. In a Phase I study involving 32 healthy volunteers not on anticoagulant therapy, AnXa proved to be safe and well tolerated.48
A placebo-controlled Phase II trial (NCT01758432) investigated the reversal effect of AnXa after dosing to steady state with direct and indirect FXa inhibitors in healthy volunteers. Boluses of AnXa ranging from 210 to 800 mg were used for reversal of FXa inhibitors.49,50 A dose-dependent reversal of the anticoagulant effect was observed when measuring anti-FXa activity with a maximum decrease of >90%.50 A subsequent infusion of 4–8 mg AnXa/min for 1–2 h preserved the reversal effect. Anti-FXa activity returned to placebo levels 2 h after the end of AnXa infusion. In parallel, plasma concentrations of unbound FXa inhibitors decreased immediately after AnXa bolus. Thrombus generation and clotting time were also dose-dependently reversed by AnXa. No thrombotic or serious adverse events were observed. However, tissue factor pathway inhibitor activity also decreased due to its binding to AnXa. These results suggest that AnXa has the potential to be a universal antidote for reversal of anticoagulation produced by direct and indirect FXa inhibitors.
In a next step, AnXa was tested in older healthy volunteers measuring anti-FXa activity. The first ANNEXA-A trial (NCT02207725) tested the reversal of apixaban 5 mg twice daily given for 4 days to 33 healthy volunteers aged 50–70 years. AnXa 400 mg i.v. bolus showed a 94% complete reversal rate, effectively and rapidly reversing the anticoagulant effect of apixaban.51 No serious adverse events or thrombotic events were observed. In the second ANNEXA-A trial, AnXa will be administered as 400 mg i.v. bolus followed by a continuous infusion of 4 mg/min for 120 min. In the ANNEXA-R trial (NCT02220725), the reversal of rivaroxaban will be tested in older healthy volunteers.52 ANNEXA-E for reversal of edoxaban is planned.
A Phase III trial will evaluate the safety and ability of AnXa in reversing the effect of Xa inhibitors in patients with an acute major bleeding episode (NCT02329327). Like RE-VERSE AD testing idarucizumab, this will be an uncontrolled, open-label study. The primary endpoint will be the proportion of patients with excellent or good haemostasis.
Ciraparantag (PER977), a small-molecule antagonist
Ciraparantag (PER977) is a synthetic, peptide-like, small molecule designed for the reversal of oral direct FXa and FIIa inhibitors, low-molecular-weight heparins, unfractionated heparin, and fondaparinux but not VKA. Given the molecular structure of PER977, immunogenic reactions are unlikely.
In ex vivo studies, PER977 completely reversed the anticoagulant activity of rivaroxaban and apixaban measured with anti-FXa activity in human plasma.53 No pro-coagulant effects were observed. In animals treated with high doses of edoxaban, PER977 reduced blood loss in bleeding models (rat tail transection).54 Similar results were observed with rivaroxaban, apixaban, and dabigatran.55,56 In parallel, PER97 restored global coagulation tests to baseline levels within 20 min. Additionally, no pro-coagulant effects were observed.55 In a liver laceration model, the highest investigated dose of PER977 and AnXa showed a similar reduction of blood loss in rabbits pretreated with rivaroxaban.57 However, PER977 did not affect anti-FXa activity in this model.
A Phase I trial (NCT01826266) evaluated the safety, tolerability, and plasma and urinary pharmacokinetics of a wide range of single i.v. doses of PER977 (5–300 mg) administered either alone or following a single dose of edoxaban.58 PER977 >100 mg reversed edoxaban 60 mg within 10 min and restored clot formation without rebound signals for 24 h.59 No pro-coagulant effects were observed. In this first trial, PER977 was safe and well tolerated.
An ongoing clinical trial (NCT02207257) evaluates the safety, tolerability, and effect on whole blood clotting time of escalating intravenous doses of PER977 (25, 50, 100, 300, and 600 mg) administered after 60 mg edoxaban as a ‘rescue’ medication in healthy volunteers and repeated for a second day to investigate any effects of PER977 on the re-anticoagulation with edoxaban and second reversal with PER977 (NCT02207257). Phase III clinical trial results need to be awaited to assess the full clinical therapeutic potential of this promising new NOAC antidote.
Areas of future research
While traditional controlled Phase III trials may not be feasible in this area of research in future, an accumulating body of evidence from observational real-world studies will be even more important.60 Areas of uncertainty to be addressed in future studies include (i) the risk of thromboembolic events in patients with immediate reversal of anticoagulation (with reversal agents without a pro-thrombotic effect per se); (ii) the net clinical benefit of specific antidotes, particularly in patients with severe but not life-threatening bleeding or urgent surgery; (iii) the effect of specific reversal agents on clinical events with specific focus on ICH; and (iv) the effect of specific reversal agents in patients with spontaneous and provoked bleeding.
Conclusions
While available clinical data for specific antidotes of NOACs are limited, it is very likely that these agents will successfully reverse the effect of NOACs as measured by specific coagulation tests. Another perceived advantage is the lack of a pro-thrombotic effect of the agent itself. However, uncontrolled studies will not give a definite answer to the question whether the clinical outcome will be improved. In the absence of specific antidotes, CFCs and/or haemodialysis for dabigatran may be a valuable alternative and may be used in addition to specific reversal agents in case of liver or renal failure. Before considering the use of a reversal agent for NOACs, a comprehensive pharmacological and clinical assessment of the usually complex situation of a critically ill patient is essential (Table 3). Furthermore, a set of general measures should precede the use of a reversal agent, as depicted in Figure 1. While the availability of specific reversal agents will increase the confidence in the benefit of NOACs in clinical practice, an overuse of specific reversal agents (beyond the tested indications) is of unclear advantage for patients. Prevention of bleeding in patients with NOACs by managing the bleeding risk of the patient and by respecting dose-reduction criteria61 and contraindications remains essential. Even in patients who have survived life-threatening bleeding, restarting anticoagulation therapy is associated with reduced risks of thromboembolic events and mortality without increasing the risk of bleeding.62 Thus, a timely restart of anticoagulant therapy (or alternative therapeutic strategies such as left atrial appendage occlusion) should be considered depending on the cause and the reversibility of the index bleeding event.
Initial assessment of patients with a major bleeding event or requiring emergency surgery
| Pharmacological considerations | Clinical considerations |
|---|---|
|
|
| Pharmacological considerations | Clinical considerations |
|---|---|
|
|
NOAC, non-vitamin K antagonist oral anticoagulant; aPTT, activated partial thromboplastin time; OAC, oral anticoagulant.
Initial assessment of patients with a major bleeding event or requiring emergency surgery
| Pharmacological considerations | Clinical considerations |
|---|---|
|
|
| Pharmacological considerations | Clinical considerations |
|---|---|
|
|
NOAC, non-vitamin K antagonist oral anticoagulant; aPTT, activated partial thromboplastin time; OAC, oral anticoagulant.
Consensus statements
– Global coagulation tests are not appropriate for measuring successful reversal of NOAC effects (ex vivo studies). Specific coagulation tests for the respective NOAC should be used.
– CFCs should be used in case of life-threatening bleeding or emergency surgery in NOAC-treated patients when a specific reversal agent is not available or there are signs of systemic coagulopathy. There is no consistent superior effect of a specific CFC (ex vivo studies and animal studies). Pro-thrombin complex concentrates should be preferred over rFVIIa based on the inherent pro-thrombotic risk.
– Idarucizumab, the first reversal agent tested in critically ill patients, is effective in reversing the effect of dabigatran measured by specific coagulation tests (uncontrolled clinical study) and has been approved by the European Medicines Agency and the U.S. Food and Drug Administration. Andexanet α targets the effect of direct and indirect FXa inhibitors and has been successfully tested in healthy volunteers so far. Ciraparantag (PER977) is designed to antagonize direct and indirect FXa inhibitors as well as dabigatran. In a Phase I study, PER977 was safe and well tolerated. Once approved by regulatory authorities, the use of specific antidotes for NOACs should be restricted to life-threatening bleeding or emergency surgery. Discontinuation of NOACs may be sufficient due to their rather short half-life for other situations requiring improved haemostasis. The effect of reversal agents on the clinical outcome in critically ill patients can only be assessed in controlled trials.
– While specific antidotes for NOACs may not have an inherent pro-thrombotic effect, the sudden termination of anticoagulation in patients with a significant pro-thrombotic risk may cause thromboembolic events (Phase III study). The time point of restarting anticoagulation may therefore be crucial for the net clinical benefit.
Authors' contributions
A.N., L.K., J.T., J.M., G.Y.H.L., S.W., and F.W.A.V.: drafted the manuscript. A.N., J.T., J.M., L.K., S.W., S.E.H., C.T.-P., K.K., B.S.L., H.D., J.C.K., D.A., R.F.S., G.Y.H.L., F.W.A.V., and S.A.: made critical revision of the manuscript for key intellectual content.
Supplementary material
Supplementary Material is available at European Heart Journal online.
Conflict of interest: A.N. reports grants, personal fees, and non-financial support from Böhringer-Ingelheim, personal fees from Pfizer, personal fees from Bristol-Myers-Squibb, personal fees, and non-financial support from Daiichi-Sankyo, personal fees from Bayer, outside the submitted work. Juan Tamargo reports grants from Astrazeneca, outside the submitted work. João Morais reports personal fees from Astrazeneca, personal fees from bayel healthcare, personal fees from BMS/Pfizer, personal fees from Lilly/Daiichi-Sankyo, personal fees from Boehringer Ingheleim, personal fees from Merck Sharp and Dhome, outside the submitted work. Lorenz Koller has nothing to disclose. Sven Wassmann reports personal fees from Bristol–Myers-Squibb, personal fees from Pfizer, personal fees from Boehringer-Ingelheim, personal fees from Bayer Healthcare, personal fees from Merck Sharp Dohme, personal fees from AstraZeneca, personal fees from Novartis, outside the submitted work. Steen Elkjær Husted reports grants from Boehringer-Ingelheim, personal fees from Boehringer-Ingelheim, personal fees from Bayer, personal fees from BMS, personal fees from Pfizer, personal fees from AstraZeneca, during the conduct of the study. Christian Torp-Pedersen reports grants and personal fees from Bayer, outside the submitted work. Keld Kjeldsen has nothing to disclose. Basil S. Lewis reports grants and personal fees from Pfizer, grants, and personal fees from BMS, grants from AstraZeneca, personal fees from Merck, outside the submitted work. Heinz Drexel reports personal fees from Bayer, personal fees from Böhringer-Ingelheim, personal fees from Pfizer, during the conduct of the study; personal fees from Bayer, personal fees from Böhringer-Ingelheim, personal fees from Pfizer, outside the submitted work. Juan Carlos Kaski reports personal fees from Sanofi, personal fees from Servier UK, personal fees from Menarini UK, outside the submitted work. Dan Atar reports personal fees from BMS/Pfizer, personal fees from Boehringer-Ingelheim, personal fees from Bayer Healthcare, outside the submitted work. Robert F. Storey reports grants, personal fees, and other from AstraZeneca, personal fees from Aspen, personal fees from PlaqueTec, personal fees from The Medicines Company, personal fees from ThermoFisher Scientific, grants and personal fees from Merck, personal fees from Correvio, personal fees from Roche, personal fees from Regeneron, personal fees from Sanofi-Aventis, personal fees and non-financial support from Accumetrics, personal fees from Daiichi-Sankyo/Eli Lilly, outside the submitted work. In addition, R.F.S. has a patent related to PEGASUS TIMI 54 study results (no financial interest) pending. Gregory Y. H. Lip has served as a consultant for Bayer, Astellas, Merck, AstraZeneca, Sanofi, BMS/Pfizer, Biotronik, Portola, and Boehringer-Ingelheim and has been on the speakers bureau for Bayer, BMS/Pfizer, Boehringer-Ingelheim, and Sanofi-Aventis, outside the submitted work. Freek W.A. Verheugt reports personal fees from Boehringer-Ingelheim, personal fees from Bristol-Myers-Squibb, personal fees from Daiichi-Sankyo, personal fees from Bayer Healthcare, during the conduct of the study. Stefan Agewall has nothing to disclose.
References
Author notes
The opinions expressed in this article are not necessarily those of the Editors of the European Heart Journal or of the European Society of Cardiology.


{kind=link}
{kind=link}